MAVE (Multiplexed Assays of Variant Effect) Data Analysis Tools
Multiplexed assays of variant effect (MAVEs) are high-throughput methods employed to investigate the impact of genetic variations in proteins or nucleic acids on their functionality.
MAVE experiments are often referred to as deep mutational scans (DMS), and offer a comprehensive and systematic approach to examining the relationship between sequence and function.
Typically, MAVE experiments involve three primary steps:
- Library generation. The first stage of a MAVE/DMS experiment involves the preparation of a library by introducing either random or specific variations across the gene or protein sequence.
- Variant selection. In the next step, the collection of mutant variants is subjected to high-throughput functional screening to assess the phenotypic consequences of the introduced variants.
- Variant scoring. During the variant scoring process, each variant is assessed based on enrichment scores within post-selected populations.
Computational tools for scoring
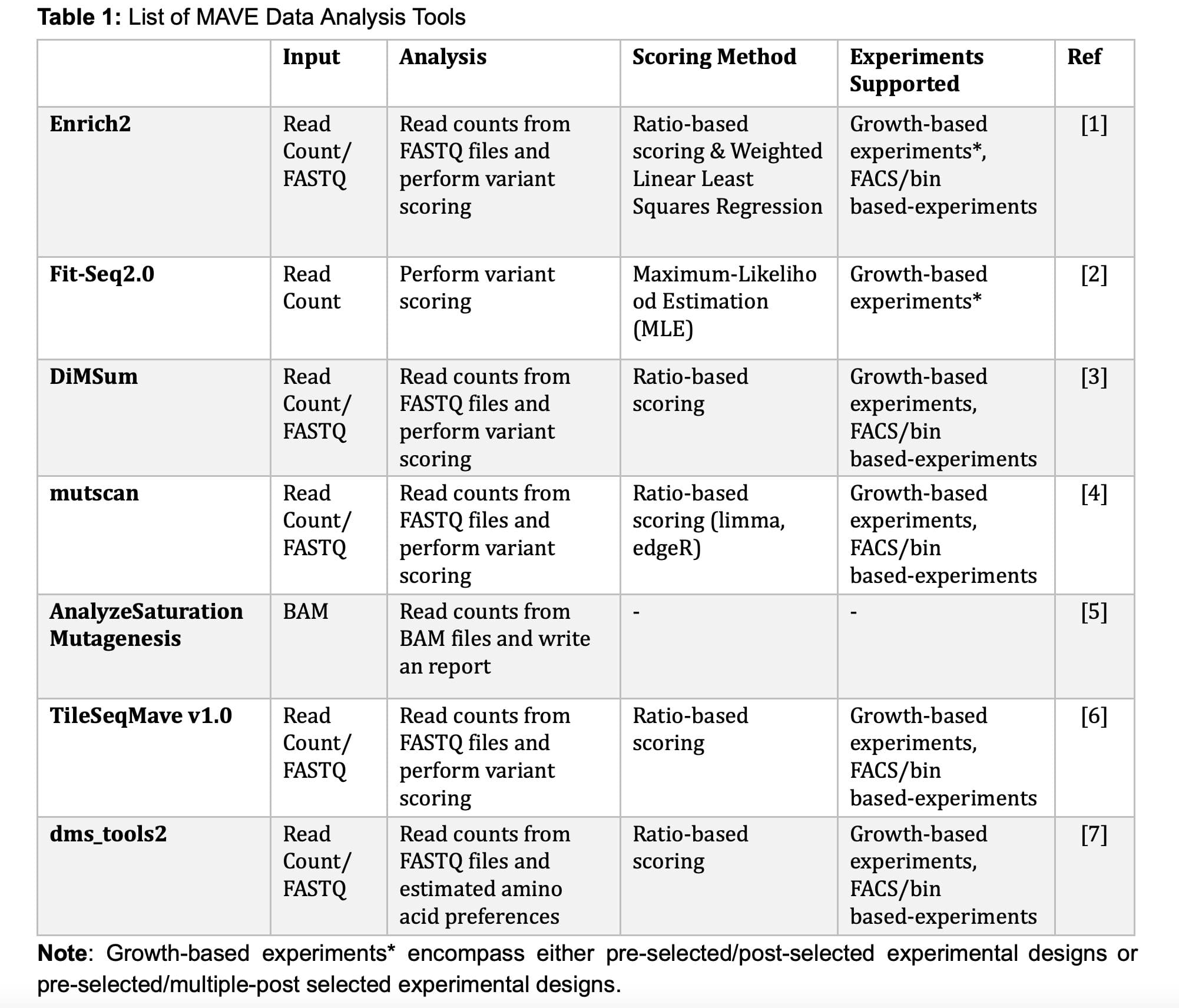
Variant scoring or analysis is a critical step in MAVE experiments, where each examined variant is assigned a score. The scoring of variants is driven by their enrichment within the post-selected populations. To ascertain variant-specific enrichment, particular sites are sequenced using next-generation sequencing technologies such as PacBio, Nanopore, and Illumina, after which variants are assessed based on their abundance in the post-selection population. As of 2023, at least seven different MAVE data analysis tools have been released (as detailed in Table 1: MAVE Data Analysis Tools). These tools are designed to extract insights from count and/or sequencing files and perform variant scoring process.
Specifically, both Enrich2 and Fit-Seq2.0 provide the capability to analyze bulk growth experiments that involve multiple timepoints within post-selected populations. In contrast, other tools, including DiMSum, mutscan, TileSeqMave v1.0, and dms_tools2, are limited to analyzing experiments with only a single pre-selected and single post-selected population.
Researchers commonly opt for TileSeqMave v1.0 when conducting MAVE experiments using a direct/tile sequence approach. The source code for TileSeqMave v1.0 is accessible in the Roth Lab's GitHub repository (https://github.com/rothlab/tileseqMave). For MAVE experiments conducted through a barcode sequence approach, researchers often prefer to use Enrich2. The source code for Enrich2 is available in the Fowler's Lab repository (https://github.com/FowlerLab/Enrich2).
Other MAVE Data Analysis Tools
Researchers employing the barcode-based approach need to incorporate an extra sequencing step called "barcode phasing" which is required to establish the associations between the barcodes and their respective variants. To effectively associate barcodes with their respective variants, various tools are at your availability, including alignparse, AssemblyPacBio, and PackRAT. Among the three tools that successfully demonstrate effective functionality, alignparse and PackRAT stand out for their comprehensive documentation, providing clear instructions on both tool operation and technical specifics.
You can find the source code for alignparse in the GitHub repository of the Bloom Lab (https://github.com/jbloomlab/alignparse), while source code for PackRAT is available in the Dunham Lab's GitHub repository (https://github.com/dunhamlab/PacRAT).
MAVE-NN is a Python package that provides a framework for generating genotype-phenotype maps from MAVE datasets [8]. Code and documentation is available at https://mavenn.readthedocs.io/en/latest/
Rererences
- Rubin, A. F., Gelman, H., Lucas, N., Bajjalieh, S. M., Papenfuss, A. T., Speed, T. P., & Fowler, D. M. (2017). A statistical framework for analyzing deep mutational scanning data. Genome biology, 18, 1-15.
- Li, F., Tarkington, J., & Sherlock, G. (2023). Fit-Seq2. 0: an improved software for high-throughput fitness measurements using pooled competition assays. Journal of Molecular Evolution, 1-11.
- Faure, A. J., Schmiedel, J. M., Baeza-Centurion, P., & Lehner, B. (2020). DiMSum: an error model and pipeline for analyzing deep mutational scanning data and diagnosing common experimental pathologies. Genome Biology, 21(1), 1-23.
- Soneson, C., Bendel, A. M., Diss, G., & Stadler, M. B. (2023). mutscan—a flexible R package for efficient end-to-end analysis of multiplexed assays of variant effect data. Genome Biology, 24(1), 1-22.
- GATK. ‘AnalyzeSaturationMutagenesis (BETA)’, 19 August 2021. https://gatk.broadinstitute.org/hc/en-us/articles/4405443726107-AnalyzeSaturationMutagenesis-BETA-.
- ‘TileSeqMave v1.0’. R. 2019. Reprint, Roth Laboratory, 10 January 2023. https://github.com/rothlab/tileseqMave.
- Bloom, J. D. (2015). Software for the analysis and visualization of deep mutational scanning data. BMC bioinformatics, 16, 1-13.
- Tareen, E., Kooshkbaghi, M., Posfai, A., Ireland, W.T., McCandlish, D.M. & Kinney, J.B. (2022). MAVE-NN: learning genotype-phenotype maps from multiplex assays of variant effect. Genome Biology, 23, 1-27
This resource was put together by Hasan Çubuk (PhD student at the University of Edinburgh) and Joseph Marsh as part of the Analysis Modelling and Prediction (AMP) workstream efforts